

Insights center
For clinical trial authorization of new drugs, sponsors must submit an IND application to the National Medical Products Administration (NMPA). To facilitate successful IND submissions, this article outlines essential documentation preparation and strategic considerations for conducting pre-IND communication meetings with the Center for Drug Evaluation (CDE), ensuring effective regulatory alignment and adequate guidance.
Regulatory Basis
According to NMPA regulations, applicants must request a communication meeting with the Drug Review Center before submitting their first clinical trial application for a new drug. This ensures subject safety and confirms the completeness of trial materials and the feasibility of the clinical study.
In 2024, CDE released guidelines to assist applicants in preparing materials for drug development and review communication meetings including meeting classification of Category I, Category II and Category III, appropriate use of these meetings and timing of meeting request.
Pre-application meetings for new drug clinical trials are classified as Category II meetings. These meetings address critical technical issues before the initial IND submission, such as whether existing data supports the trial and if subject risks are manageable. A written response or meeting is typically provided within 60 working days after the application.
Application Materials Preparation
1. Communication Meeting Application Form:
Drug development basics: Applicant details, drug name, chemical/structure (for Chinese medicine), proposed use, dosage form, route/method of administration, and development strategy.
Meeting specifics: Content required for new drug clinical trial pre-application meetings.
Meeting type: Category II clinical trial pre-application meeting.
Meeting classification: clinical trial pre-application meeting.
Meeting format: face-to-face, video, telephone or written response (select one).
Purpose: brief description (e.g., assess if existing research data supports the proposed clinical trial).
Proposed dates: For in-person, video, or phone meetings, propose 3 dates within 60 working days post-application.
Meeting agenda: Provide a brief schedule (meetings ≤1 hour).
Participants: Names and roles.
2. Meeting Deck
PPT outlines drug development progress and key discussion points. Recommended content:
Company & Drug Overview: Summarize company background, development timeline, and current status.
Study Data Summary: Highlight key pharmacological, non-clinical, and clinical findings supporting the trial.
Discussion Topics: Discipline-specific questions (e.g., pharmacology, toxicology, clinical trial design/statistics). Include brief R&D context and rationale for each topic.
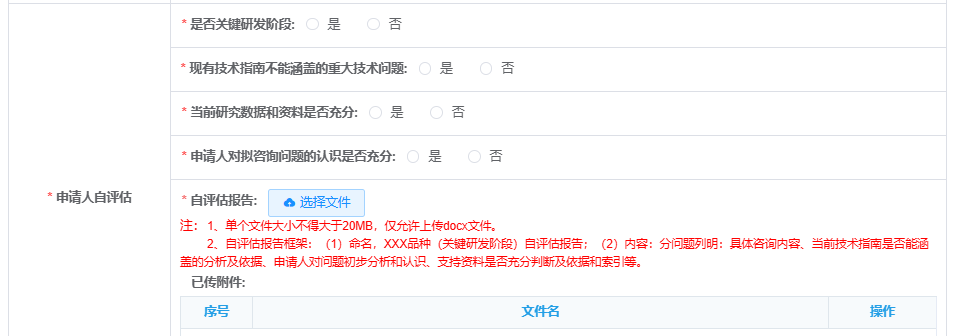
3. Self-Assessment Report (Mandatory since June 2024):
CDE requires applicants to submit a self-assessment with communication meeting requests. This report evaluates:
Whether the topic is critical to R&D progress.
If it addresses gaps in current guidelines.
Adequacy of supporting data.
Applicant’s preparedness to discuss the issue.
Purpose: Ensure meetings focus on essential, well-defined topics.
4. Submission of supporting materials
In addition to the three core components (application form, PPT, self-assessment report), applicants must submit supporting materials necessary to validate the feasibility and safety of the drug development and clinical trial design. When applying via CDE’s "Applicant Window," questions must be categorized by discipline, with each category requiring a dedicated list of up to 10 questions. Each question should include a brief background explanation, its purpose, and the applicant’s proposed position.

Supporting materials vary based on the issue type and may include:
1. A quality overview or pharmacological summary must include key research findings, supporting rationale, and proposed solutions for discussion. For biologics, this entails providing critical pharmacology-toxicology data, representative batch samples of the original solution and formulation, and evidence of pharmacological comparability. Safety analyses should cover biological activity, immunogenicity, and specific risks (e.g., exogenous factors, tumorigenicity), alongside quality characterization and preliminary method validation. For preventive vaccines, include details on the bacterial/viral strains used in production, cell matrices, and third-party verification plans/results to ensure safety and efficacy.
2. A non-clinical research review must summarize pharmacology and toxicology data in supporting the proposed clinical trial. This includes the research strategy, completed pharmacodynamics, pharmacokinetics, and toxicology results aligned with the clinical plan, particularly evidence validating the drug’s efficacy for the target indication. Key details should cover species selection rationale for toxicity testing, identified safety risks (e.g., NOAEL), and justification for the initial human trial dose based on preclinical findings.
3. Clinical trial materials (e.g., risk management plans, investigator manuals, informed consent forms) must demonstrate alignment between pharmacology, non-clinical/preclinical data (if available) and key trial design elements, such as justifying the starting dose, dose-escalation rationale, and safety risk assessments for first-in-human studies.
4. Submit R&D context, supporting data/literature, and proposed solutions for discussion topics.
Precautions
1. Before submitting the application, ensure all information is complete, accurate, and standardized. The submitted questions should address the applicant’s key challenges in drug development and clinical trials, supported by a clear problem background, relevant data, proposed solutions, and a risk assessment. This ensures effective communication and resolution of critical issues before trial initiation.
2. Questions should be structured as "whether" (yes/no) questions, not open-ended ones.
3. Ensure all submitted data is well-documented, as the data may be referenced in the formal IND application.
4. Time Planning: As a Category II meeting, the preclinical trial communication meeting requires a 60-working-day response time. Post-feedback IND submission adjustments may be needed, so integrate this timeline into overall registration planning.
5. Stay Updated: Track CDE's latest meeting requirements, guidelines and system updates to ensure compliance.
Conclusion
China's pre-clinical trial communication meeting is critical for ensuring smooth drug development and reliable clinical data. Proper preparation enables effective CDE discussions and valuable feedback, supporting subsequent trial success and regulatory approval.
SDM offers comprehensive market access solutions, including regulatory strategy, clinical development planning, and registration pathway optimization for global pharma, biotech, and medtech companies. Our core team brings over 20 years of global registration expertise, with deep knowledge of multi-regional regulatory frameworks and proven success in aligning product development programs with health authority requirements through effective agency communication. Contact us to discuss your regulatory needs.
The application process and requirements are subject to change based on CDE's latest policies. For the most current and accurate information, please refer to CDE's official website or authorized announcements.
This session focuses on Stakeholder Engagement: developing strategies for collaborative project decision-making and execution.
The updated draft guidance involves a focus on clinical trial design, regulatory considerations, and whether these trials can demonstrate that the drugs can maintain weight loss as determined by BMI.
Let's take a look back at the previous two installments of the Clinical Data Management “PM” series: Scope Management, which clarifies the scope of responsibilities of all parties involved in a clinical trial, and Project Resource Management, which focuses on the utilization of company and personal resources to accomplish data management tasks. In this installment, we will focus on the most important part of project management - project schedule management - to share the timeline planning and progress follow-up of data management activities, so as to efficiently complete the data management work under the premise of ensuring the data quality and reaching the important milestones of the project.
In the last session, we learned about project scope management for data management work, identifying the scope of responsibilities for data cleansing and data management activities. After defining the scope of the data management work, in this session, we will learn how to mobilize the resources within the scope of work to carry out the data management work more efficiently and with higher quality.
Shanghai SDM Vaccine Data Management Department, in collaboration with the International Project Department, has launched a series of training sessions on "Application of Project Management Knowledge in Data Management." The Application of Project Management Knowledge in Data Management Work contains eight modules, including Project Integration Management, Project Scope Management, Project Progress Management, Project Quality Management, Project Resource Management, Project Communication Management, Project Risk Management, and Project Stakeholder Management, etc. It mainly refers to the theoretical knowledge of the Guide to the Project Management Body of Knowledge (PMBOK Guide) and combines the content of the data management work and practical experience of the project.
Want to quickly penetrate the Chinese, American and European pharmaceutical markets? Registering for communication exchanges is the key!
SDM PV team detects drug safety risks.
SDM Vaccine Experts Share Roadmap to Avoid Clinical Trial Pitfalls.
this article outlines essential documentation preparation and strategic considerations for conducting pre-IND communication meetings with CDE, ensuring effective regulatory alignment and adequate guidance.
Global Vaccine Solutions via Multidimensional Strategies.
July 23, 2025 GSK disclosed that the FDA has postponed PDUFA date of the Blenrep® (belantamab mafodotin) combination therapy BLA. The agency established a new action date of October 23, 2025 for completion of BLA review.
SDM Bioservices has successfully established a hybrid immuno-capture LC-MS/MS platform for the simultaneous quantification of ADC total antibody, conjugated antibody, conjugated drug, and free small-molecule payload. This approach significantly reduces reliance on specific antibody reagents, enables rapid method development and validation, and supports high-throughput sample analysis—thereby accelerating project timelines and advancing drug development efforts.
Premier Li Qiang has signed a State Council decree, promulgating the "Regulations on the Administration of Clinical Research and Translation of Novel Biomedical Technologies." This important regulation was adopted at the State Council executive meeting on September 12, 2025, and will take effect on May 1, 2026. This establishes a comprehensive legal framework for China's oversight of novel biomedical technologies throughout the entire chain from research to application.
Authored by the SDM Bioanalysis Team, this article delves into the principles, delivery systems and advantages of mRNA technology, provides a comprehensive overview of its applications and clinical breakthroughs in three major fields—infectious disease prevention, cancer therapy and protein replacement therapy—and elaborates on the key bioanalytical methodologies.
Get in touch with SDM experts for your questions or comments and a member of our team will get back to you directly.
Let's Start a Conversation


